
Introduction the FDA Recall Process Dexcom Device Recall: The FDA Recall Process
Welome to this authoritative guide in the Dexcom Device Recall in which we will demystify the U.S. Food and Drug Adminsitration (FDA) recall process. Dexcom devices sit at the center of day-to-day diabetes management for many patients, caregivers, and clinicians. That reality makes any potential Dexcom device recall a high attention event, not only because of the technology involved, but because of the downstream effects on dosing decisions, alarms, trend interpretation, and clinical confidence.
At the same time, “recall” is often misunderstood. In the United States, most medical device recalls are not a single dramatic moment where a regulator removes a product from the market. Instead, a recall is typically a structured corrective action process. It is often initiated by the manufacturer, overseen by the FDA and executed through field actions that can include software updates, customer notifications, product corrections, and in some cases removals.
This article explains how the FDA recall process works for medical devices, how a Dexcom related recall would typically progress from detection to closure, what recall classifications mean, and what practical steps patients and providers can take to manage risk proactively.
If you or a loved one used a defective Dexcom device and suffered harm when your Dexcom device malfunctioned, contact Dexcom Recall Lawsuit Lawyer Timothy L. Miles for a free case evaluation today as you may be eligible for a Dexcom Device Recall Lawsuit and potentially be entitled to substantial compensation. (855) 846–6529 or [email protected].

What “Device Recall” Means in FDA Terms
In FDA usage, a medical device recall is a firm’s method of removing or correcting marketed devices that the FDA considers to be in violation of laws it enforces, and which may pose a risk to health. The key word is correcting. For digital and connected products like Dexcom’s, a recall can be satisfied by a correction that meaningfully reduces risk.
Such risks could arise from defective Dexcom devices, which may necessitate recalls. A recall can involve several actions such as:
- Updating device firmware or mobile application software.
- Modifying labeling, instructions for use, or training materials.
- Replacing a component or accessory.
- Adjusting manufacturing controls or acceptance criteria.
- Requiring customer checks, configuration changes, or workflow changes.
A recall is therefore less about stigma and more about governance. It is a structured, documented process that aims to reduce risk quickly, communicate clearly, and verify effectiveness.
Who Initiates a Dexcom Recall: FDA vs Manufacturer
A common misconception is that the FDA “issues” most recalls. In practice, most recalls are voluntary actions initiated by the manufacturer after internal detection, customer complaints, post market signals, supplier findings, or verification failures.
The FDA’s role is to:
- Assess risk and determine the appropriate recall classification.
- Review the firm’s recall strategy, including who is notified and how.
- Monitor execution through status reports, audit checks, and effectiveness verification.
- Publicly communicate recall information through FDA databases and safety communications when appropriate.
- Close the recall once the agency is satisfied that corrective actions were implemented and verified.
The FDA can also request or mandate action in certain circumstances, but the operational work of notifying customers, distributing corrections, and tracking responses is typically led by the manufacturer.
The Typical FDA Recall Lifecycle (How It Works End to End)
Although each event is unique, FDA device recalls tend to follow a repeatable sequence. Understanding the sequence helps patients and clinicians interpret what they are seeing when recall notices appear in public databases or customer emails.
1) Signal Detection and Triage
Signals can come from many sources:
- Customer complaints about performance, alarms, connectivity, or accuracy behavior.
- Adverse event reports (including reports submitted to FDA databases).
- Internal quality system findings, such as nonconformances or out of specification results.
- Supplier quality issues affecting sensors, adhesives, transmitters, chargers, or packaging.
- Software defect discovery through internal testing, cybersecurity monitoring, or field analytics.
The firm performs triage to determine whether the issue is:
- A non safety matter (handled as routine support or minor change),
- A field correction that does not meet recall thresholds, or
- A recall requiring formal regulatory reporting and structured execution.
This phase is where risk is framed. The central question is not “Is there a bug?” but “Could this reasonably contribute to patient harm, and how quickly do we need to reduce risk?”
2) Health Hazard Evaluation (HHE) and Risk Assessment
A Health Hazard Evaluation is a formal risk analysis that estimates the probability and severity of harm if the issue occurs. For a continuous glucose monitoring (CGM) ecosystem, hazards might include:
- Missed hypoglycemia alerts due to alarm or notification failures.
- Inaccurate glucose readings leading to incorrect insulin dosing decisions.
- Data gaps that affect therapy decisions or clinician review.
- Adhesion failures contributing to early sensor loss and loss of trending.
- App display or unit conversion issues that could be misinterpreted.
- Interoperability issues with other devices or systems.
This risk framing matters because it often drives:
- The recall classification (Class I, II, or III),
- The urgency of notifications,
- The scope of affected lots or software versions,
- The required effectiveness checks.
3) Recall Decision and Regulatory Notification
If the firm concludes the field action meets recall criteria, it typically notifies the FDA and submits recall information, including:
- Product identification (model numbers, software versions, lot numbers).
- Distribution scope (where and when shipped).
- Affected population and estimated number of units.
- Description of the failure mode and hazards.
- Proposed corrective action and customer instructions.
- Communication plan and timeline.
The FDA reviews the submission, may request clarifications, and then posts recall information in its public recall database once the event is entered and processed.
If you or a loved one used a defective Dexcom device and suffered harm when your Dexcom device malfunctioned, contact Dexcom Recall Lawsuit Lawyer Timothy L. Miles for a free case evaluation today as you may be eligible for a Dexcom Device Recall Lawsuit and potentially be entitled to substantial compensation. (855) 846–6529 or [email protected].
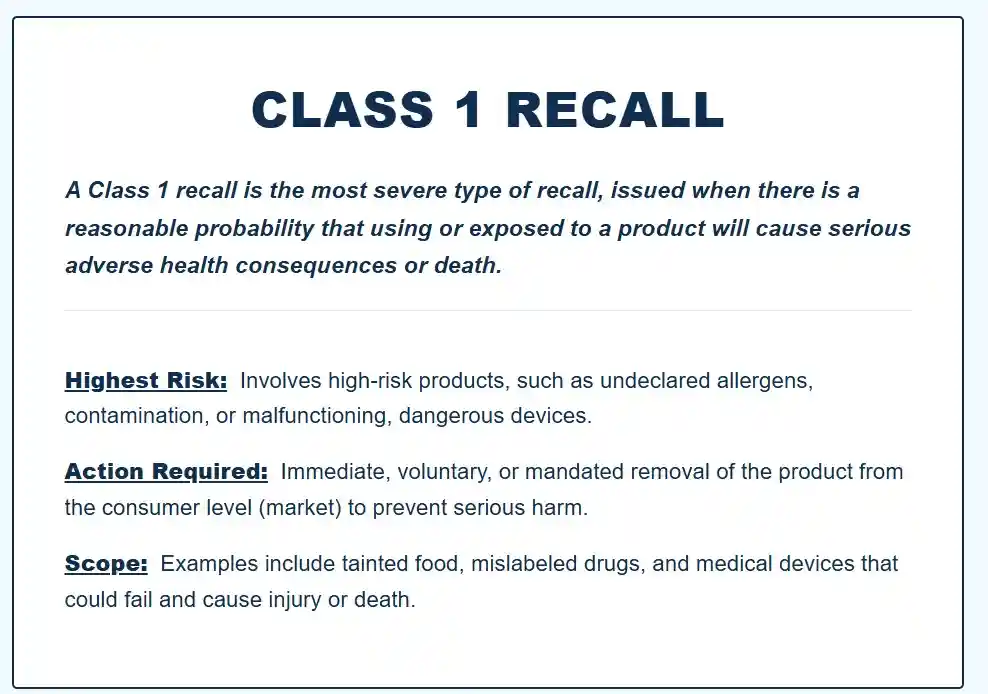
4) Recall Classification: Class I, Class II, Class III
The FDA assigns a classification based on the health risk posed by the product defect or violation.
- Class I recall: A reasonable probability that the use of, or exposure to, the product will cause serious adverse health consequences or death.
- Class II recall: Use of, or exposure to, the product may cause temporary or medically reversible adverse health consequences, or the probability of serious adverse consequences is remote.
- Class III recall: Use of, or exposure to, the product is not likely to cause adverse health consequences.
For CGM related products, classification depends on the specific failure mode, the likelihood of occurrence, and the extent to which users can detect the problem before harm occurs. A defect that is obvious to the user may be lower risk than one that silently degrades performance.
Classification is not a moral judgment about the company. It is a standardized FDA risk label intended to drive appropriate urgency and response.
5) Recall Strategy and Customer Communications
A recall strategy defines:
- Depth of recall: consumer level, retail level, or healthcare provider level.
- Public warning: whether broad public communication is needed.
- Effectiveness checks: how the firm will verify that customers received and understood the communication.
Dexcom recall communications, when they occur, often involve digital channels because many users are app based:
- Email notifications to registered users.
- In app alerts or banners directing users to instructions.
- Website recall pages with affected identifiers and FAQs.
- Communication to distributors, pharmacies, and durable medical equipment suppliers.
- Outreach to clinics and health systems that prescribe or support the product.
From a governance perspective, the key is clarity. A high quality recall notice should state plainly:
- What is affected.
- What the risk is.
- What the user must do now.
- How to identify if their specific device or lot is impacted.
- What replacement, update, or remedy is offered.
- How to contact support.
6) Corrective Actions: Correction vs Removal
The FDA recall framework distinguishes between:
- Correction: Repair, modification, adjustment, relabeling, or inspection of a product without physical removal from its point of use.
- Removal: Physical removal of the product from its point of use, often returning it to the firm or disposing of it.
In modern CGM ecosystems, many recall remedies are corrections, such as:
- Updating the mobile app to address an alarm handling defect.
- Updating transmitter firmware if supported.
- Updating labeling to clarify use conditions, compatibility, or warning statements.
- Replacing a subset of sensors tied to a manufacturing lot.
The FDA cares less about which category is chosen and more about whether the remedy reduces risk promptly and verifiably.
7) Effectiveness Checks and Recall Status Reports
Once communications are issued, the firm conducts effectiveness checks to confirm that the right audience received the message and took the required action. Methods can include:
- Confirmation emails or response tracking.
- Distributor acknowledgments and inventory reconciliations.
- Sample calls to users or clinics.
- Return rates or update adoption metrics for software based corrections.
The firm also submits periodic recall status reports to the FDA describing:
- Number of customers notified.
- Quantity corrected or removed.
- Any issues with implementation.
- Results of effectiveness checks.
- Adjustments to strategy if needed.
8) Recall Termination (Closure)
A recall remains open until the FDA is satisfied that:
- The firm made reasonable efforts to notify all affected parties.
- The corrective action addressed the hazard.
- The action was effective and appropriately verified.
- The product in the field has been corrected, removed, or otherwise controlled.
Recall closure is important from a systems standpoint because it indicates the formal end of the field action, not necessarily that the product category is “risk free.” All medical devices remain subject to ongoing post market surveillance as per the FDA’s postmarket requirements.
How to Interpret a Dexcom Recall Notice When You See One
When a recall notice appears in the FDA database or in a Dexcom communication, users often ask one question first: “Does this apply to me?”
To answer it, focus on identifiers rather than headlines. Relevant identifiers may include:
- Sensor lot numbers and expiration dates.
- Transmitter model numbers.
- Receiver model numbers.
- App version numbers and supported operating systems.
- Distribution dates and regions.
A recall may apply only to a narrow slice of distribution, such as a limited lot range produced within a specific time window, or a particular software version released before a patch.
From a safety standpoint, precision matters. Broad anxiety does not improve outcomes. Accurate identification and compliance with the corrective action does.
FDA Recall Databases and Where to Check (Without Guesswork)
If you are monitoring for Dexcom related recall activity, rely on primary sources:
- FDA Medical Device Recalls database: the authoritative listing of device recalls and classifications.
- Dexcom official support and product notices: manufacturer instructions and FAQs.
- Healthcare organization safety notices: some clinics and hospitals redistribute relevant recall communications.
Be cautious with secondary sources that summarize recall activity without listing exact affected identifiers. In recall management, details are the control.
Official Support Channels
- Technical Support (24/7): 1-844-607-8398 for troubleshooting and sensor replacements.
- General Inquiries/Orders: 1-888-738-3646 (Mon-Fri, 6 am – 5 pm PST).
- Online Support/Chat: Access via My Dexcom Account.
- Dexcom CARE: Free virtual educational sessions.
What to Do If You’re Affected by a Dexcom Recall
If you find yourself affected by a Dexcom recall, it’s crucial to follow the manufacturer’s instructions carefully. However, if you suffer any harm as a result of using a recalled product, you may have legal options available to you. For instance, you could potentially seek compensation in a Dexcom recall lawsuit which can help cover medical expenses or other damages incurred due to the faulty device.
For more information on how to navigate through such situations, you can refer to Health Canada’s medical devices recall guide.
If you or a loved one used a defective Dexcom device and suffered harm when your Dexcom device malfunctioned, contact Dexcom Recall Lawsuit Lawyer Timothy L. Miles for a free case evaluation today as you may be eligible for a Dexcom Device Recall Lawsuit and potentially be entitled to substantial compensation. (855) 846–6529 or [email protected].
Why CGM Recalls Can Look Different Than Traditional Device Recalls
CGM is a hybrid product category. It includes disposable components, reusable electronics, and software. That structure changes recall execution.
Software as a Recall Vector
If a defect is corrected through an app update, the recall becomes partly a deployment problem:
- How quickly does the update reach users?
- Do users have compatible operating systems?
- Are automatic updates enabled?
- Are there enterprise managed phones in clinical environments?
Effectiveness checks in these cases often focus on adoption rates and confirmation that the corrected version is installed. This highlights the importance of change management in medical devices, which can play a significant role in ensuring successful recall execution.
Interoperability and Ecosystem Dependencies
CGM products can interact with:
- Smartphones and operating system updates.
- Smartwatches and notification frameworks.
- Cloud services and data sharing functions.
- Third party apps or connected insulin delivery systems, where applicable.
A recall may therefore reference compatibility or specific configurations. From a governance perspective, this expands the “device” boundary. Risk management must account for ecosystem behavior, not just the sensor.

Human Factors and Use Conditions
In CGM, user behavior is part of the safety case. A recall may instruct users to:
- Confirm alarms are enabled and audible.
- Verify units of measure.
- Perform confirmatory fingerstick testing when symptoms do not match readings.
- Replace sensors that show specific failure patterns.
These instructions are not blame shifting. They are part of risk control, particularly when a defect can be mitigated through user checks while a replacement or update is deployed. This underscores the necessity for developing a comprehensive medical device recall strategy to reduce patient safety risks.
What Patients Should Do If They Suspect Their Dexcom Product Is Affected
If you believe your Dexcom component, app, or accessory might be subject to a recall or correction, a structured response is more effective than improvisation.
- Identify the product and version
- Record the sensor lot number, transmitter model, receiver model, and app version. Take screenshots where relevant.
- Check official recall instructions
- Compare your identifiers to the affected identifiers listed by Dexcom and in the FDA recall entry.
- Follow the corrective action exactly
- If the remedy is an app update, update promptly and confirm the version installed. If the remedy is replacement, follow the replacement process and keep records.
- Use clinically appropriate backup practices
- If a recall notice recommends confirmatory testing under certain conditions, follow that guidance and consult your clinician for individualized advice.
- Report problems through the right channel
- Notify Dexcom support for product performance issues and consult your healthcare team for clinical concerns. Adverse events can also be reported through FDA channels, but manufacturer support is often the fastest path for troubleshooting and replacement logistics.
The operational goal is continuity of care. The governance goal is traceability, meaning you can demonstrate what you received, what you did, and when you did it.
What Clinics, Diabetes Educators, and Health Systems Should Do
Healthcare organizations should treat CGM recalls as a predictable governance event, not an ad hoc fire drill. Effective programs are repeatable, auditable, and patient centered.
Key actions include:
- Recall intake and triage: route notices to clinical engineering, pharmacy and supply chain, informatics, and diabetes care leadership as appropriate.
- Patient segmentation: identify patients on the affected devices, versions, or lots using available records.
- Standardized outreach scripts: provide consistent instructions and minimize conflicting guidance.
- Documentation: record outreach attempts, patient acknowledgments, and clinical recommendations.
- Escalation pathways: define when to schedule urgent visits, adjust therapy plans, or advise temporary alternative monitoring.
This is where proactive governance becomes measurable. The organizations that perform best are those with established recall playbooks, defined ownership, and consistent communication.
The Regulatory and Quality Controls Behind Recalls (Why They Matter)
Recalls sit inside broader regulatory obligations, including:
- Quality System Regulation (QSR) requirements for design controls, production and process controls, corrective and preventive action (CAPA), complaint handling, and purchasing controls.
- Medical Device Reporting (MDR) obligations for reporting certain adverse events and malfunctions.
- Post market surveillance practices that track real world performance.
A recall is often the visible tip of a larger corrective action program. Strong governance means strong documentation, strong root cause analysis, strong corrective action design, and strong verification that the fix works.
Repetition is valuable here. Governance requires documentation. Governance requires verification. Governance requires follow through.
Common Misinterpretations That Create Unnecessary Panic
Several patterns appear repeatedly in recall news cycles:
- “Recall” equals “device is unusable.”
- Not necessarily. Many recalls involve corrections or specific lots. The remedy may be straightforward.
- “Class I” equals “the product will harm you.”
- Class I indicates a higher potential risk category, but it does not mean harm is inevitable. It means urgency and compliance matter more.
- “If I did not receive a notice, I am not affected.”
- Possibly, but not guaranteed. Users change emails, apps, and phone numbers. Checking identifiers is the reliable method.
- “Social media summaries are sufficient.”
- Recall management depends on exact identifiers and exact instructions. Summaries often omit both.
A Practical Framework for Future Readiness
Dexcom users, clinicians, and organizations can reduce disruption from future recall events through a few proactive habits:
- Keep device identifiers accessible: lot numbers, model numbers, app versions.
- Maintain current contact information in your Dexcom account.
- Enable app updates where clinically appropriate and confirm versions after major releases.
- Establish a backup monitoring plan with your clinician for periods of device interruption.
- Use official channels for recall verification and instructions.
This is not fear-based preparation. It is continuity planning. It is governance at the individual level, aligned with governance at the organizational level.
Conclusion
A Dexcom device recall, when it occurs, should be understood as a structured safety and compliance mechanism, not as a single event defined by headlines. The FDA recall process is built around classification, communication, correction, verification, and closure. It is built to reduce risk quickly. It is built to improve accountability. It is built to protect patients.
For patients and providers, the practical priority is straightforward: verify whether you are affected using identifiers, follow the corrective action precisely, document what you did, and consult a clinician when clinical decisions are impacted.
In some cases, patients may need to consider legal options if they are adversely affected by these recalls. For instance, Do You Qualify for a Dexcom Lawsuit in 2026? could be a pertinent question to explore.
Proactive governance is not optional in connected healthcare. It is the foundation for trust, the mechanism for integrity, and the pathway to safer innovation in 2026 and beyond.
If you or a loved one used a defective Dexcom device and suffered harm when your Dexcom device malfunctioned, contact Dexcom Recall Lawsuit Lawyer Timothy L. Miles for a free case evaluation today as you may be eligible for a Dexcom Device Recall Lawsuit and potentially be entitled to substantial compensation. (855) 846–6529 or [email protected].
Frequently Asked Questions about the Dexcom Device Recall
What does a Dexcom device recall mean according to the FDA?
A Dexcom device recall, as defined by the FDA, is a manufacturer’s method of removing or correcting marketed devices that may pose a health risk or violate laws enforced by the FDA. It often involves corrective actions such as software updates, labeling changes, component replacements, or customer notifications to reduce risk rather than just product removal.
Who typically initiates a Dexcom device recall and what role does the FDA play?
Most Dexcom device recalls are voluntary actions initiated by the manufacturer after detecting issues through customer complaints, internal findings, or supplier problems. The FDA oversees these recalls by assessing risk, reviewing recall strategies, monitoring execution, communicating publicly when needed, and closing the recall after verifying corrective actions.
What are common steps involved in the FDA’s medical device recall process for Dexcom devices?
The typical FDA recall lifecycle includes signal detection and triage from various sources like customer complaints and internal testing; conducting a Health Hazard Evaluation (HHE) to assess risk severity and probability; making a recall decision with regulatory notification; executing field actions such as software updates or product corrections; and finally verifying effectiveness before closing the recall.
What types of risks can lead to a Dexcom device recall?
Risks prompting a Dexcom device recall can include missed hypoglycemia alerts due to alarm failures, inaccurate glucose readings affecting insulin dosing decisions, data gaps impacting therapy review, adhesion failures causing early sensor loss, app display errors leading to misinterpretation, and interoperability issues with other medical devices or systems.
How does a Dexcom device recall impact patients and clinicians in diabetes management?
A Dexcom device recall can significantly affect day-to-day diabetes management by influencing dosing decisions, alarm reliability, trend interpretation, and clinical confidence. Patients and clinicians need clear communication about any corrective actions like software updates or usage changes to manage risks proactively while maintaining effective glucose monitoring.
What practical steps should patients take if their Dexcom device is subject to a recall?
Patients should follow all instructions provided by Dexcom regarding the recall, stay informed through official communications including FDA databases and company notifications, consult their healthcare provider for personalized advice, implement any recommended software updates or usage modifications promptly, and report any ongoing issues to ensure safety during diabetes management.